Graphical Fragment Assembly GFA 2.0
Shaun Jackman @sjackman
2017-01-18
https://sjackman.ca/gfa2-slides

Shaun Jackman
BC Cancer Agency Genome Sciences Centre
Vancouver, Canada
@sjackman | github.com/sjackman | sjackman.ca

Common file format
- Interoperable modular tools
- Deduplication of effort
- Data visualization
Alignment
- Alignment and variant calling is a pipeline of two stages
- Alignment reads FASTQ produces SAM/BAM
- Variant calling reads SAM/BAM produces VCF
Benefits
Things we take for granted
- Mix and match aligners and variant callers
- Support tools (samtools) shared by everyone
- Visualization tools (IGV) shared by everyone
- Troubleshooting intermediate stages
- All possible because of SAM/BAM
Assembly
- Assembly is a pipeline
Overlap, Layout, Consensus - Common input file format (FASTQ)
- Common output file format (FASTA)
- No common intermediate file format
Deduplication of effort
- Developing a new variant caller doesn't require developing a new aligner
- Developing a new assembler shouldn't require developing a new read overlap tool
GFA 1
- Started as a blog post by Heng Li
- Sequence overlap graph
- Vertices (segments) are contigs
- Edges (links) are their overlaps
- Tab separated format
- Extensible (optional tagged fields)
Implementations
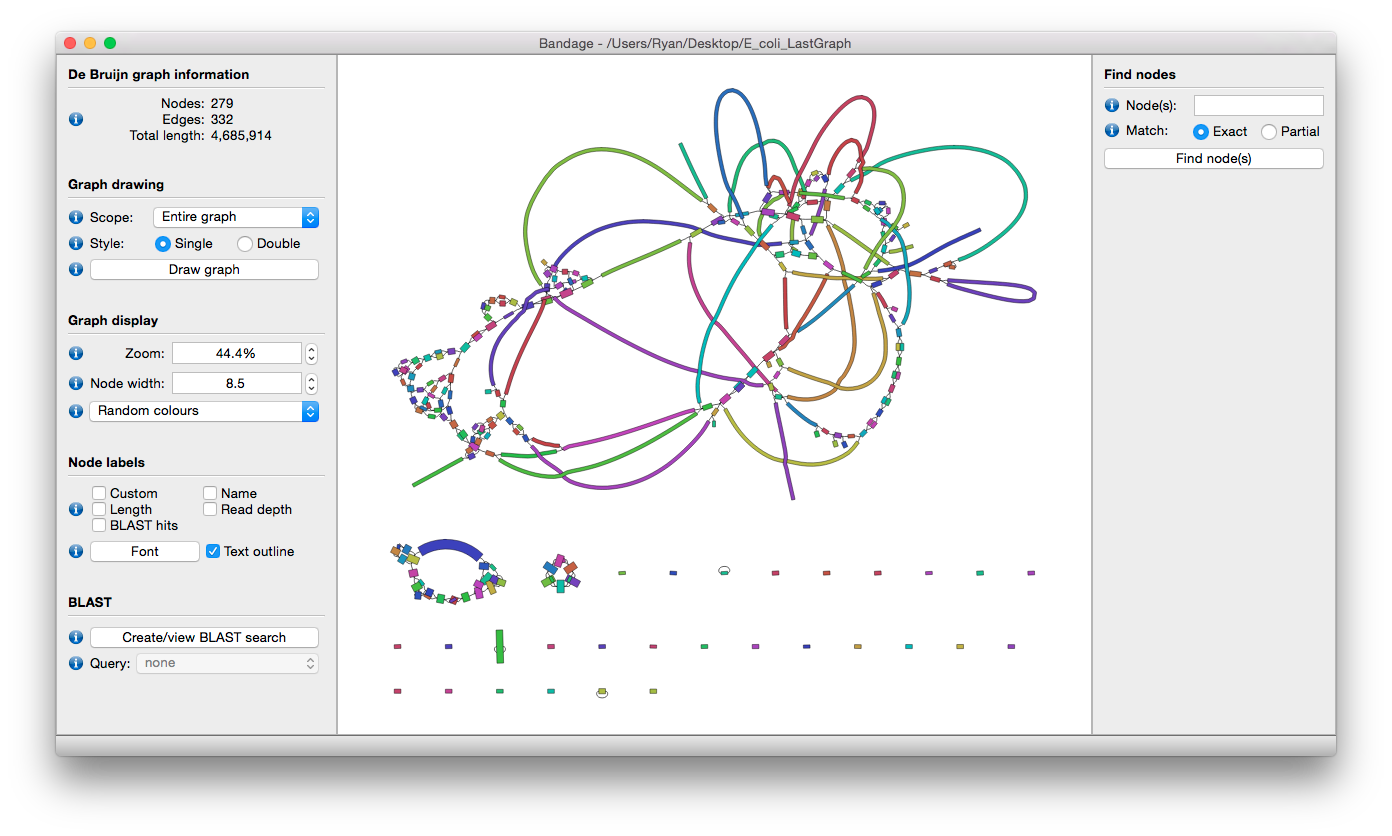
Limitations
- Intended for
- Short read assembly
- Contig overlap graph (not reads)
- CIGAR unwieldy for long read alignment
- Links represent only dove-tail overlaps
GFA 2
- Short and long read assembly
- Read overlap graph or contig overlap graph
- Arbitrary alignments (not just dovetail)
- Correcting reads
- Identifying repeats
- CIGAR or compact trace alignments
- Gap edges and paths for scaffolding
Exciting Possibilities
- Mixing components of assembly pipelines
- Visualization of intermediate results
aids troubleshooting - Align reads to a pan-genome
- Graph-aware gene annotation
- Innovation in modular assembly tools
- Identify heterozygous contigs
- Identify misassembled contigs
- Modular scaffolding tools
- One tool produces gap edges
- Another tool creates scaffolds
xkcd: Standards

fin
Shaun Jackman
BC Cancer Agency Genome Sciences Centre
Vancouver, Canada
@sjackman | github.com/sjackman | sjackman.ca
GFA Specification
https://github.com/GFA-spec/GFA-spec
Slides
https://sjackman.ca/gfa2-slides
Markdown source code
https://github.com/sjackman/gfa2-slides